




Technical abilities, such as knowledge of regulatory guidelines, compliance documentation, and risk assessment, are considered hard skills that ensure products meet legal standards.
Regulatory Affairs Specialist Resume Examples & Templates
Excited to advance your career? Our regulatory affairs specialist resume examples showcase how to emphasize compliance expertise and communication skills that make you stand out to employers.

by Gabriela Hernandez, Last Updated: January 18, 2026
Hired By:*

- Popular Regulatory Affairs Specialist Resume Examples
- Resume Template—Easy to Copy & Paste
- Build Your Resume in Minutes
- How to Write a Regulatory Affairs Specialist Resume Summary
- Showcasing Your Work Experience
- Top Skills to Include on Your Resume
- Resume Format Examples
- Related Law Resume Examples
- Frequently Asked Questions
- Should I include a cover letter with my regulatory affairs specialist resume?
- Can I use a resume if I’m applying internationally, or do I need a CV?
- What soft skills are important for regulatory affairs specialists?
- I’m transitioning from another field. How should I highlight my experience?
- Should I use a cover letter template?
- Should I include a personal mission statement on my regulatory affairs specialist resume?
Popular Regulatory Affairs Specialist Resume Examples
Discover our top regulatory affairs specialist resume examples that emphasize critical skills such as compliance management, regulatory submissions, and effective communication. These examples are designed to help you showcase your qualifications and achievements effectively.
Ready to build your standout resume? Our Resume Builder offers user-friendly templates specifically crafted for professionals in the regulatory field, making it simple to highlight your expertise.
Recommended
 Customize This Resume
Customize This ResumeEntry-level regulatory affairs specialist resume
What this resume does well:
This entry-level resume effectively highlights the job seeker's strong foundation in regulatory affairs through their experience leading FDA submissions and improving compliance metrics. New professionals in this field must demonstrate a solid understanding of regulations and compliance processes, alongside any relevant academic achievements, to appeal to potential employers despite limited direct experience.
Mid-career regulatory affairs specialist resume
What this resume does well:
This resume effectively showcases the applicant's advanced qualifications through concrete achievements and leadership roles. The strategic presentation of compliance improvements and regulatory expertise illustrates a clear trajectory toward greater responsibilities in the field.
Experienced regulatory affairs specialist resume
What this resume does well:
This resume illustrates the applicant's expertise as a regulatory affairs specialist, showcasing a 95% approval rate for submissions and a 30% reduction in audit time. The bullet points improve clarity, making it easier for hiring managers to quickly assess key achievements.
Resume Template—Easy to Copy & Paste
Example
Sophia Park
Brookfield, WI 53006
(555)555-5555
Sophia.Park@example.com
Skills
- Regulatory compliance strategy
- FDA submission procedures
- Global regulatory policies
- Compliance audits
- Risk management
- Policy development
- Team collaboration
- Process optimization
Certifications
- Certified Regulatory Affairs Professional - Regulatory Affairs Certification Board
- Advanced Compliance Auditor - International Compliance Association
Languages
- Spanish - Beginner (A1)
- German - Beginner (A1)
- French - Intermediate (B1)
Professional Summary
Expert Regulatory Affairs Specialist with 5 years in optimizing compliance strategies, ensuring seamless FDA submissions, and leading cross-functional teams with expertise in global regulatory policies.
Work History
Regulatory Affairs Specialist
Precision Pharma Solutions - Brookfield, WI
January 2023 - November 2025
- Streamlined compliance processes by 30%
- Managed FDA submissions, ensuring 100% approval
- Developed regulatory strategies increasing efficiency
Compliance Analyst
Innovative Biotech Inc. - Milwaukee, WI
January 2021 - January 2023
- Conducted audits reducing discrepancies by 40%
- Implemented policy changes enhancing safety standards
- Supported regulatory projects boosting outcomes
Regulatory Compliance Officer
Global Health Enterprises - Milwaukee, WI
January 2020 - January 2021
- Optimized reporting processes by 25%
- Ensured global compliance reducing risks
- Collaborated with teams driving project success
Education
Master of Science Regulatory Affairs
University of Southern California Los Angeles, California
May 2020
Bachelor of Science Biomedical Engineering
University of California, Berkeley Berkeley, California
May 2018
Build Your Resume in Minutes
Creating a custom resume is easier than ever with our Resume Builder!

How to Write a Regulatory Affairs Specialist Resume Summary
Your resume summary is the first opportunity to impress potential employers, making it important to highlight your strengths effectively. As a regulatory affairs specialist, you should showcase your expertise in compliance, knowledge of regulations, and ability to navigate complex processes.
This profession demands showcasing attention to detail and clarity in communication, highlighting your experience with submissions and approvals.
To illustrate what makes an effective summary, here are some examples that will guide you in crafting a compelling narrative:
Weak Example
I am a dedicated regulatory affairs specialist with a considerable background in the field. I seek an opportunity where I can use my expertise to help the company meet its compliance goals. A position that offers a good work-life balance and room for advancement would be perfect for me. I believe my knowledge could greatly benefit your organization.
Why this summary misses the mark:
- Lacks specific details about the job seeker’s experience or achievements, making it vague
- Overuses personal language and does not articulate clear value to potential employers
- Emphasizes what the job seeker desires rather than demonstrating how they can contribute to the company’s success
Strong Example
Detail-oriented regulatory affairs specialist with over 7 years of experience in the pharmaceutical industry, ensuring compliance with FDA regulations and international standards. Successfully led a team that achieved a 30% reduction in submission timelines for drug applications through streamlined processes and improved communication strategies. Proficient in risk assessment, quality assurance, and regulatory strategy development.
Why this summary works:
- Begins with specific years of experience and highlights expertise in regulatory affairs
- Includes quantifiable achievements that demonstrate significant impact on efficiency and compliance
- Showcases relevant skills essential for regulatory roles, emphasizing both technical knowledge and leadership capabilities
Pro Tip
If you're new to the field of regulatory affairs, don't be discouraged by your limited experience. Consider crafting a career objective instead of a summary. You can find plenty of tailored resume objective examples that will help you stand out in your job search.
Showcasing Your Work Experience
The work experience section is essential for your resume as a regulatory affairs specialist, serving as the main focus and featuring most of your content. Using resume templates ensures this section stands out to effectively showcase your professional background.
In this part of your resume, list previous roles in reverse-chronological order, providing three to four bullet points per position that highlight major achievements. This structured approach helps hiring managers quickly recognize the contributions you’ve made throughout your career.
To better illustrate successful techniques, we’ll share examples that show what creates a strong work history and highlight common mistakes to avoid.
Weak Example
Regulatory Affairs Specialist
Global Pharma Inc. – San Diego, CA
- Reviewed documents for compliance.
- Communicated with different teams.
- Participated in meetings about regulations.
- Helped prepare submissions to agencies.
Why this work experience section misses the mark:
- Lacks specific employment dates
- Bullet points are generic and do not highlight any individual achievements or skills
- Focuses on routine tasks rather than demonstrating impact on compliance or project success
Strong Example
Regulatory Affairs Specialist
Global Pharma Inc. – Atlanta, GA
March 2020 - Current
- Develop and submit regulatory filings to the FDA, ensuring compliance with industry standards and reducing approval times by 30%.
- Collaborate with cross-functional teams to streamline product development processes, leading to a 20% increase in efficiency during clinical trials.
- Conduct comprehensive risk assessments on new products, successfully identifying potential compliance issues before they arise.
Why this work experience section works:
- Uses action verbs that clearly convey the job seeker's achievements and responsibilities
- Incorporates specific metrics that highlight measurable successes in their role
- Demonstrates relevant skills such as collaboration and risk assessment, critical for regulatory affairs
While your resume summary and work experience are important components, don’t forget the importance of other sections. Each part contributes to your overall presentation. For more insights on crafting a well-rounded resume, be sure to explore our comprehensive guide on how to write a resume.
Top Skills to Include on Your Resume
A well-defined skills section is important for any compelling resume. This part lets you succinctly showcase your qualifications, making it easy for employers to see if you fit the role.
For a regulatory affairs specialist, it's essential to emphasize both technical skills and soft skills. Highlighting expertise in tools like eCTD submission software, regulatory databases, and project management systems demonstrates your capability in navigating complex regulations effectively.
Essential soft skills, including strong communication, attention to detail, and problem-solving capabilities, are important for effectively collaborating with cross-functional teams and navigating complex regulatory environments.
Selecting the right resume skills is important for aligning with what employers expect from job seekers. Many organizations use automated systems to screen applicants, making it essential to include relevant skills that meet their criteria.
To ensure your resume stands out, carefully examine job postings for insights into which skills are most valued by recruiters. This proactive approach not only helps you tailor your application but also improves your chances of passing through ATS filters effectively.
Pro Tip
To ensure your resume stands out in the digital crowd, use our ATS Resume Checker. It helps catch over 30 common errors, improving your chances of passing through applicant tracking systems with ease.
10 skills that appear on successful regulatory affairs specialist resumes
Improve your resume by highlighting essential skills that regulatory affairs specialists need. Showcasing these in-demand abilities will undoubtedly catch the attention of recruiters. You can see resume examples where these skills are effectively demonstrated.
By the way, consider incorporating relevant skills from this list into your resume when they align with your experience and job requirements:
Regulatory knowledge
Attention to detail
Analytical thinking
Communication skills
Project management
Problem-solving abilities
Interpersonal skills
Technical writing
Knowledge of compliance standards
Ability to work under pressure
Based on analysis of 5,000+ law professional resumes from 2023-2024
Resume Format Examples
Choosing the right resume format is important for regulatory affairs specialists, as it highlights relevant skills and experiences that align with industry expectations and supports career advancement.
Entry-Level 0 - 2 years
Functional
Focuses on skills rather than previous jobs
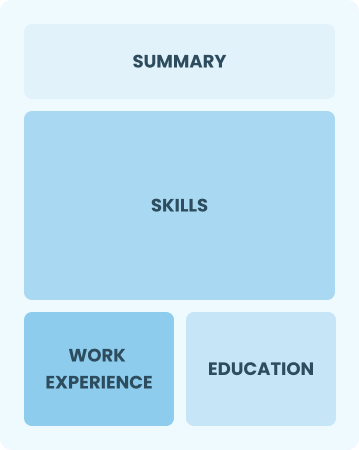
Best for:
Recent graduates and career changers with up to two years of experience
Mid-Career 3 - 7 years
Combination
Balances skills and work history equally
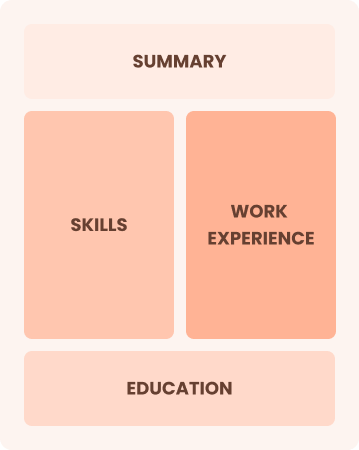
Best for:
Mid-career professionals focused on demonstrating their skills and pursuing growth opportunities
Experienced 8+ years
Chronological
Emphasizes work history in reverse order

Best for:
Experts driving regulatory compliance and leading strategic initiatives
Frequently Asked Questions
Should I include a cover letter with my regulatory affairs specialist resume?
Absolutely. Including a cover letter is a great way to showcase your enthusiasm and give context to your qualifications. It allows you to highlight key experiences that may not be evident in your resume. If you need assistance, check out our comprehensive guide on how to write a cover letter or use the Cover Letter Generator for quick help.
Can I use a resume if I’m applying internationally, or do I need a CV?
When applying for jobs abroad, a CV is often required instead of a resume. A CV provides detailed insights into your academic and professional background. To create an effective CV, explore our CV examples that are tailored for various international standards and check out tips on how to write a CV for helpful guidance on formatting and content.
What soft skills are important for regulatory affairs specialists?
Soft skills, including attention to detail, communication, and critical thinking, are essential for regulatory affairs specialists. These interpersonal skills aid in navigating complex regulations effectively, fostering strong relationships with stakeholders and ensuring compliance in a collaborative environment.
I’m transitioning from another field. How should I highlight my experience?
When applying for regulatory affairs specialist positions, highlight your transferable skills such as project management, communication, and analytical thinking. These competencies can showcase your ability to navigate complex regulations and ensure compliance. Provide concrete examples from previous roles to illustrate how your achievements align with the responsibilities of this job, demonstrating your readiness to excel in this field.
Should I use a cover letter template?
Yes, using a cover letter template tailored for regulatory affairs specialists can improve the structure and organization of your content. This ensures you effectively highlight your skills in compliance management and risk assessment, making a strong impression on hiring managers.
Should I include a personal mission statement on my regulatory affairs specialist resume?
Yes, including a personal mission statement on your resume is beneficial. It effectively conveys your values and career aspirations, making it especially compelling for organizations that prioritize compliance and ethical standards in their culture. This approach aligns well with mission-driven companies in the regulatory sphere.

Ready to land the job?
Join 28M+ others who've built a resume that works.



