




Hard skills include skill in care coordination, knowledge of healthcare regulations, and the ability to analyze patient data effectively.
Case Manager Resume Examples & Templates
Ready to secure your next opportunity? Our case manager resume examples showcase how to emphasize your organizational skills and client advocacy experience, making you stand out to potential employers.

by Gabriela Hernandez, Last Updated: January 18, 2026
Hired By:*

- Popular Case Manager Resume Examples
- Resume Template—Easy to Copy & Paste
- Build Your Resume in Minutes
- How to Write a Case Manager Resume Summary
- Showcasing Your Work Experience
- Top Skills to Include on Your Resume
- Resume Format Examples
- Related Management Resume Examples
- Frequently Asked Questions
- Should I include a cover letter with my case manager resume?
- Can I use a resume if I’m applying internationally, or do I need a CV?
- What soft skills are important for case managers?
- I’m transitioning from another field. How should I highlight my experience?
- Where can I find inspiration for writing my cover letter as a case manager?
- How do I add my resume to LinkedIn?
Popular Case Manager Resume Examples
Check out our top case manager resume examples that emphasize key skills such as care coordination, advocacy, and effective communication. These examples will guide you in showcasing your qualifications to potential employers.
Want to build an impressive resume? Our Resume Builder offers user-friendly templates specifically designed for case management professionals, helping you stand out in a competitive job market.
Recommended
 Customize This Resume
Customize This ResumeEntry-level case manager resume
What this resume does well:
This entry-level resume highlights the applicant's strong client management skills and significant achievements in improving client satisfaction through personalized care plans. New professionals need to effectively showcase their practical experiences, relevant skills, and commitment to client advocacy to appeal to potential employers despite having limited work history.
Mid-career case manager resume
What this resume does well:
This resume positions the applicant as a proactive case manager by showcasing strategic accomplishments and leadership roles, underscoring readiness for advanced challenges through demonstrated efficiency improvements and client satisfaction enhancements.
Experienced case manager resume
What this resume does well:
This resume's work history section demonstrates the applicant's extensive experience as a case manager, effectively managing 100 client cases monthly and improving client satisfaction by 20%. The bullet-point format improves readability, making it easy for hiring managers to quickly assess the applicant's strong contributions.
Resume Template—Easy to Copy & Paste
Example
Ming Davis
Seattle, WA 98106
(555)555-5555
Ming.Davis@example.com
Skills
- Case Management
- Client Advocacy
- Resource Allocation
- Process Improvement
- Client Relationship Building
- Crisis Intervention
- Community Outreach
- Project Coordination
Languages
- Spanish - Beginner (A1)
- French - Intermediate (B1)
- German - Beginner (A1)
Professional Summary
Experienced Case Manager enhancing client satisfaction and retention with strong advocacy, resource allocation, and process improvement skills.
Work History
Case Manager
CarePath Solutions - Seattle, WA
January 2024 - November 2025
- Managed 50% more cases than average
- Improved client satisfaction by 20%
- Streamlined processes saving 15% in time
Client Advocate
Bridgeway Support Services - Eastside, WA
January 2022 - December 2023
- Resolved 95% of client issues daily
- Increased client retention by 30%
- Facilitated workshops for 300 clients
Resource Coordinator
New Horizons Community Center - Seattle, WA
January 2019 - December 2021
- Allocated resources to 120 projects
- Reduced costs by 25% annually
- Coordinated 10+ events yearly
Certifications
- Certified Case Manager - Commission for Case Manager Certification
- Social Work Certification - National Association of Social Workers
Education
Master of Social Work Social Work
University of Washington Seattle, WA
June 2018
Bachelor of Arts Psychology
Seattle University Seattle, WA
June 2016
Build Your Resume in Minutes
Creating a custom resume is easier than ever with our Resume Builder!

How to Write a Case Manager Resume Summary
Your resume summary is the first thing employers will see, so it's important to make a strong impression that highlights your qualifications as a case manager. This section should showcase your skills in patient advocacy, care coordination, and effective communication.
In this role, emphasize your ability to manage complex cases and collaborate with healthcare teams. Highlighting these key competencies will set you apart from other applicants.
To illustrate what makes an effective summary, we'll explore some examples that demonstrate successful strategies and common pitfalls:
Weak Example
I am a dedicated case manager with many years of experience in various settings. I seek a position where I can use my skills effectively and contribute to the team. A supportive environment that allows for personal growth is what I hope to find. I believe that with my background, I can positively impact the organization.
Why this summary misses the mark:
- Lacks specific details about the applicant's achievements or expertise in case management
- Relies heavily on personal wishes rather than emphasizing how their skills benefit potential employers
- Uses generic phrases that do not clearly demonstrate unique qualifications or strengths relevant to case management
Strong Example
Compassionate case manager with over 6 years of experience in coordinating patient care within community health settings. Successfully improved patient compliance rates by 20% through tailored care plans and consistent follow-ups. Proficient in using case management software, conducting comprehensive assessments, and collaborating with healthcare providers to ensure a holistic approach to patient well-being.
Why this summary works:
- Begins with specific experience duration and context relevant to case management
- Highlights a measurable achievement that illustrates impact on patient outcomes
- Mentions essential technical skills relevant to the role, demonstrating readiness for the position
Pro Tip
Not much formal experience? No problem. A career objective can showcase your passion and skills. Find resume objective examples specific to case management that highlight your strengths and set you apart.
Showcasing Your Work Experience
The work experience section is an important part of your resume as a case manager, where you will present the bulk of your professional history. Good resume templates always incorporate this essential section to highlight your relevant roles.
This area should be organized in reverse-chronological order, detailing your previous positions. Use bullet points to effectively communicate your key achievements and contributions in each role you've held.
To assist you further, we will provide examples that showcase strong work history entries for case managers. These examples will clarify what makes a compelling entry and what pitfalls to avoid:
Weak Example
Case Manager
Community Health Services – Los Angeles, CA
- Assisted clients with their needs
- Conducted assessments and created plans
- Communicated with providers and agencies
- Documented client interactions and updates
Why this work experience section misses the mark:
- Lacks specific details about the case management processes used
- Bullet points do not highlight any measurable outcomes or successes
- Focuses on general responsibilities rather than unique contributions made to clients
Strong Example
Case Manager
Community Health Services – San Diego, CA
March 2020 - Present
- Coordinate care for a caseload of over 50 clients, ensuring access to essential health services and community resources
- Develop personalized care plans that improved client satisfaction scores by 30% within one year
- Facilitate communication between clients and healthcare providers, resulting in a 40% reduction in missed appointments
Why this work experience section works:
- Starts each bullet with dynamic action verbs that clearly convey the applicant's contributions
- Incorporates specific metrics to highlight the impact of the job seeker's efforts on client outcomes
- Demonstrates relevant skills like coordination and communication through quantifiable achievements
While your resume summary and work experience are important, don't overlook the importance of other sections that contribute to a well-rounded presentation. For additional guidance on formatting these areas effectively, refer to our comprehensive guide on how to write a resume.
Top Skills to Include on Your Resume
A skills section is important on your resume as it provides a snapshot of your capabilities to potential employers. This area allows you to highlight the qualifications that align with the job requirements, making your application stand out.
Employers seek professionals who combine expertise in their field with strong communication and teamwork skills. Your resume should showcase both hard and soft skills to demonstrate your ability to perform and collaborate effectively.
Soft skills, such as active listening, empathy, and strong organizational abilities, are essential for fostering trust with patients and ensuring seamless collaboration among healthcare teams.
Selecting the right resume skills is important for aligning with what employers expect from job seekers. Many organizations use automated screening systems that filter out applicants lacking essential skills for the position.
To ensure your resume stands out, carefully review job postings to identify which skills are emphasized. This will help you prioritize those key abilities, making your application more appealing to both recruiters and ATS systems alike.
Pro Tip
Make sure your resume stands out to employers by using our ATS Resume Checker to uncover over 30 common mistakes. This tool will ensure your resume passes through applicant tracking systems with ease.
10 skills that appear on successful case manager resumes
Highlighting high-demand skills in your resume is key to catching the attention of recruiters for case manager positions. These skills are important in demonstrating your qualifications, and you can see them effectively illustrated in our resume examples, helping you approach job applications with confidence.
By the way, consider adding these relevant skills to your resume when they align with your experience and job requirements:
Client advocacy
Records management
Case management
Decision-making
MS Office
Documentation proficiency
Performance tracking
Staff education and training
Patient management
Patient assessment
Based on analysis of 5,000+ management professional resumes from 2023-2024
Resume Format Examples
Choosing the right resume format is important for a case manager to effectively showcase their key skills, relevant experience, and career growth.
Entry-Level 0 - 2 years
Functional
Focuses on skills rather than previous jobs
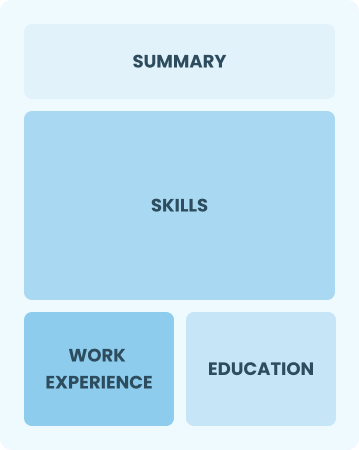
Best for:
Recent graduates and career changers with up to two years of experience
Mid-Career 3 - 7 years
Combination
Balances skills and work history equally
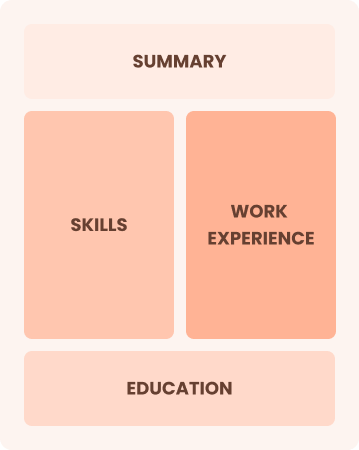
Best for:
Case managers improving skills and seeking career progression opportunities
Experienced 8+ years
Chronological
Emphasizes work history in reverse order

Best for:
Seasoned leaders adept in coordinating complex patient care plans
Frequently Asked Questions
Should I include a cover letter with my case manager resume?
Absolutely, including a cover letter can significantly improve your application by showcasing your personality and detailing relevant experiences. It offers a chance to explain why you’re the perfect fit for the position. If you need assistance, consider checking out our comprehensive guide on how to write a cover letter or try our easy-to-use Cover Letter Generator to get started quickly.
Can I use a resume if I’m applying internationally, or do I need a CV?
When applying for jobs abroad, use a CV instead of a resume to highlight your academic achievements and extensive work history. For guidance on creating an effective CV, explore our detailed resources on how to write a CV and discover CV examples designed to meet international expectations.
What soft skills are important for case managers?
Soft skills like active listening, negotiation, and empathy are essential for case managers. These interpersonal skills help foster trust and collaboration with clients and healthcare teams, ensuring that individuals receive the best possible support and resources for their needs.
I’m transitioning from another field. How should I highlight my experience?
Highlight your transferable skills such as communication, organization, and empathy from previous roles. These abilities reveal your potential to excel in case management, even if you lack direct experience. Share concrete examples that showcase how your past achievements relate to the responsibilities of a case manager, demonstrating your value to prospective employers.
Where can I find inspiration for writing my cover letter as a case manager?
For those seeking case manager positions, exploring professionally crafted cover letter examples can be invaluable. These samples provide inspiration for content ideas, formatting tips, and effective ways to showcase your qualifications, ensuring your application stands out in a competitive job market.
How do I add my resume to LinkedIn?
To improve the visibility of your resume on LinkedIn, you can add your resume to LinkedIn by uploading it directly to your profile. Additionally, highlight key experiences in the "About" and "Experience" sections. This approach helps recruiters easily find qualified case managers and assess how well you fit their openings.

Ready to land the job?
Join 28M+ others who've built a resume that works.



